}
Fig. 1: Amino Acid Sequence (260 aa) of N-acetylneuraminic Acid (2gfh) protein from Mus musculus (House Mouse)
A total of 500 proteins were yielded.
Only a 38 proteins were selected for comparison as these had shown higher homology to the query sequence, in contrast with the remainder of the search results.
These proteins were chosen according to their bit scores and E-values. Two more outlier partial sequences contributing to poor overall alignment (huge deletion gaps) were subsequently removed. The remaining 36 sequences were used for the generation of the phylogenetic tree (and bootstrapped tree as well).
The bacteria sequence matches used for multiple sequence analysis and phylogenetic tree mapping were representative of the other baterial sequences not selected. These selected sequences had the highest bit scores and E-values.
Multiple Sequence Alignments (msa)
See Materials and Methods
The following sequence alignment was obtained (Fig. 2)
From the alignments, gi|10888xy and gi|10888yz are representative of gi|108881764 and gi|108881765 respectively. Both these hypothetical proteins belong to the mosquito Aedes aegypti.
The identifier numbers for these two proteins were initially changed to an alpha-numeric one, due to the inability of Phylip to generate a tree from the original identifiers. This was due to the fact that the programme only took the first five numeric digits (10888), thereby resulting in a programme error prompt which listed both proteins as duplicates (from the identifier numbers). Both these identifiers were subsequently renamed for the final phylogenetic tree.
The topmost sequence belongs to the query protein.
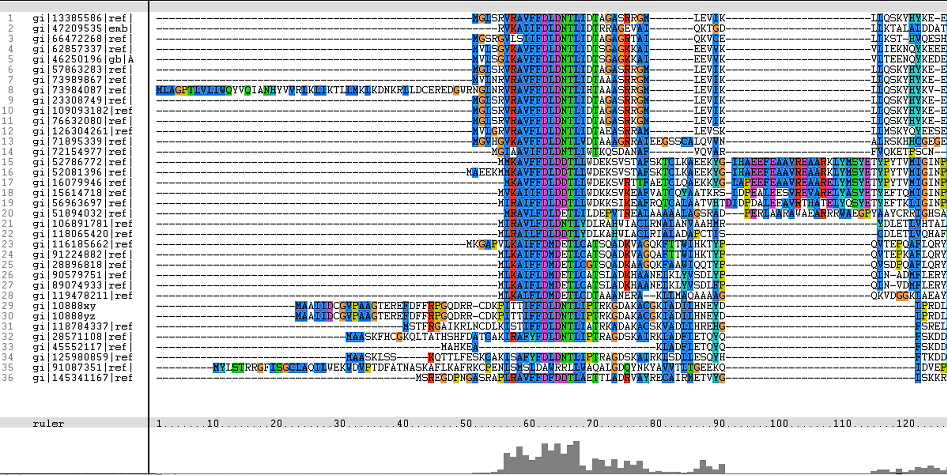
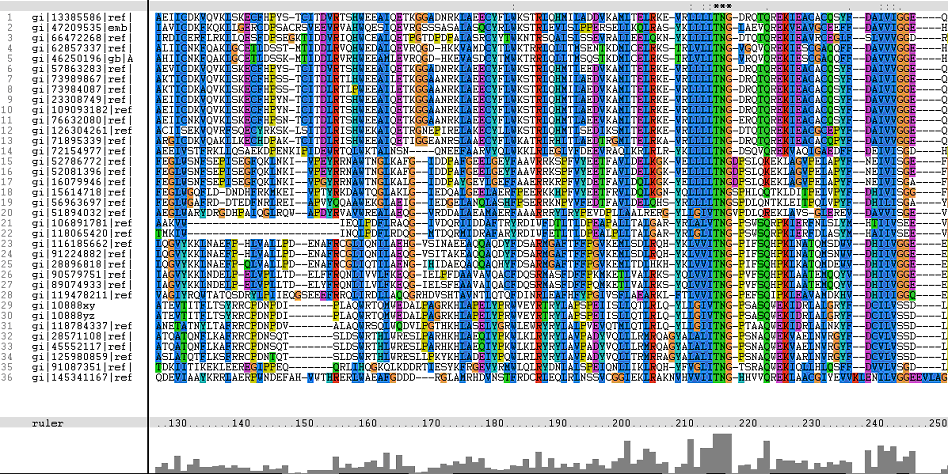
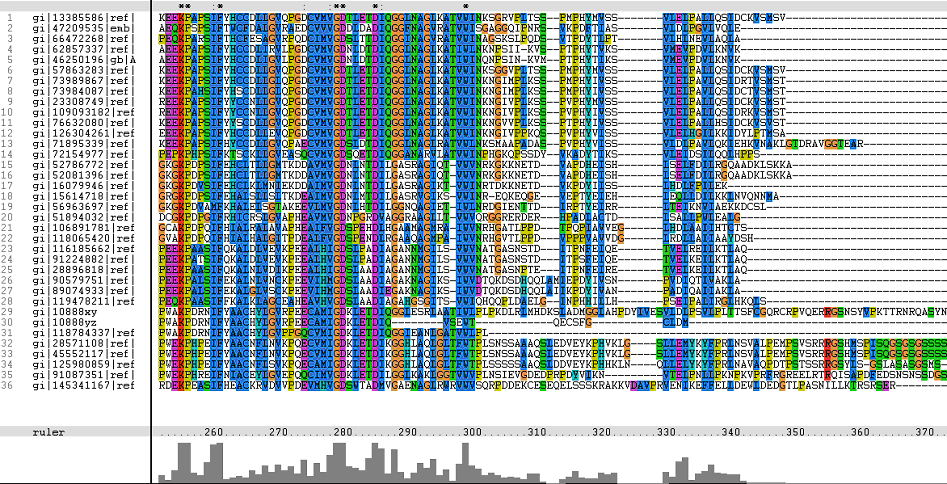
Figure 1. Multiple Sequence Alignments of 2gfh amino acid sequences with that of other similar protein sequences in various organisms.
From the multiple sequence alignment, it can be observed that there are domain conservations throughout the protein sequences. Small insertion and deletion gaps were noticeable along the alignment as well. A particularly large insertion gap was observed between amino acids 91 to 114.
The organisms with the large insertion gaps were as identified below:
- Bacillus licheniformis
- Bacillus subtilis
- Bacillus halodurans
- Bacillus clausii
- Symbiobacterium thermophilum
All five sequences belong to bacteria. With the exception of Symbiobacterium thermophilum, the rest are Bacillus sp.
Symbiobacterium are an uncultivable thermophiles isolated from compost. Their survival based mainly on microbial commensalism. This bacteria can only grow in vitro, if it is co-cultured with Bacillus sp. bacteria. This could therefore explain its genetic association with Bacillus, as observed from the sequence alignment.
However, interestingly, Bacillus is classified as Gram-positive, while Symbiobacterium is a Gram-negative bacterium. As observed from the sequence alignment, other Gram-negative bacterium protein sequences (Vibrio Sp.) do not contain the large gap insertion at the 91 to 114 amino acid positions, with the exception of Symbiobacterium. Hence, more genetic (and even functional) analysis might be necessary to determine the hydrolase protein relationship between the Gram-positive Bacillus with the Gram-negative Symbiobacterium.
A highly conserved (with invariant) section of amino acids (LV)–(LVA)–(LIV)–(LIV)-T-N-G was observed in all the sequences from amino acid 211 to 217 in the alignment. Downstream of this conserved portion of genes are 5 more invariant positions (1 or 2 amino acids in length).
From these short conservation regions, the functions or even structure of the encoded proteins could have significance in its evolutionary pattern.
Phylogenetic Map (Tree)
See Materials and Methods
The tree was plotted to obtain the phylogenetic lineage (Fig. 3):
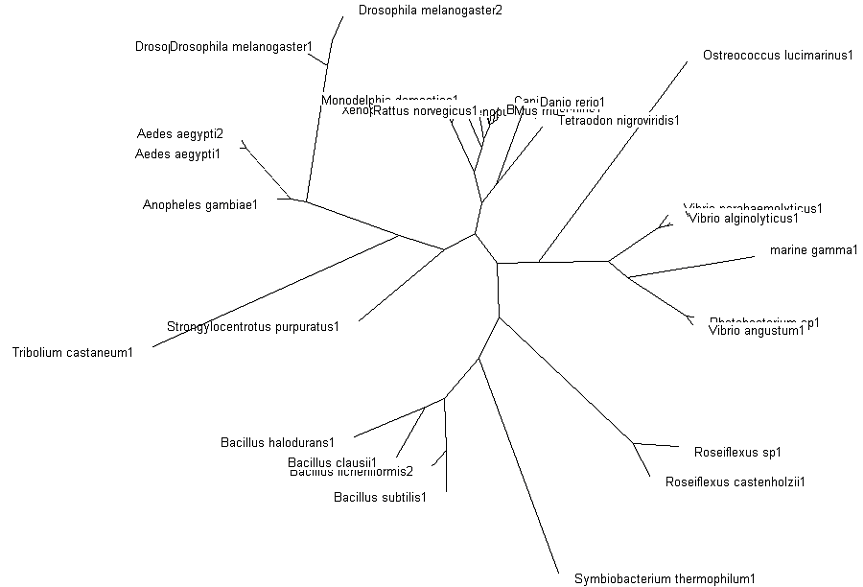
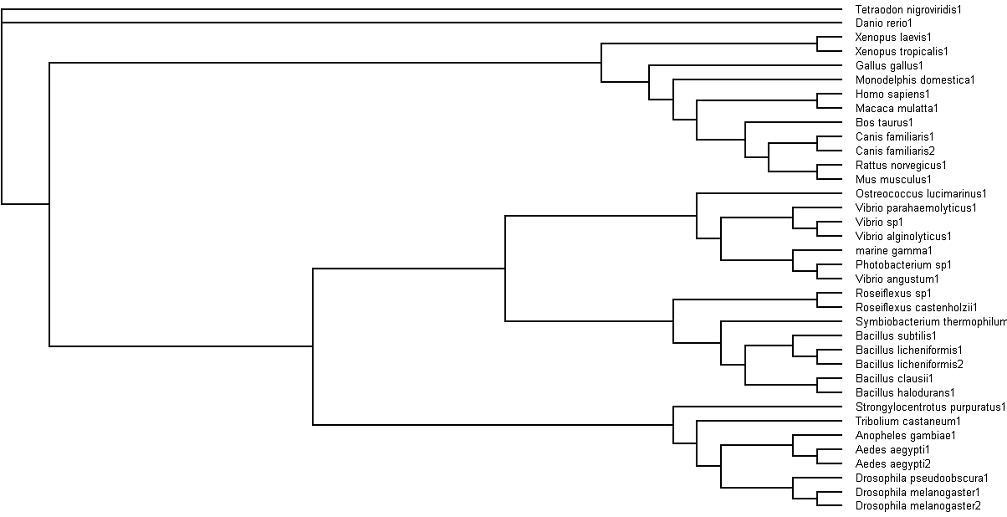
Fig. 3: Phylogenetic tree showing organisms with related protein sequence homology in Radial Tree view (top); and Rectangular Cladogram view (bottom)
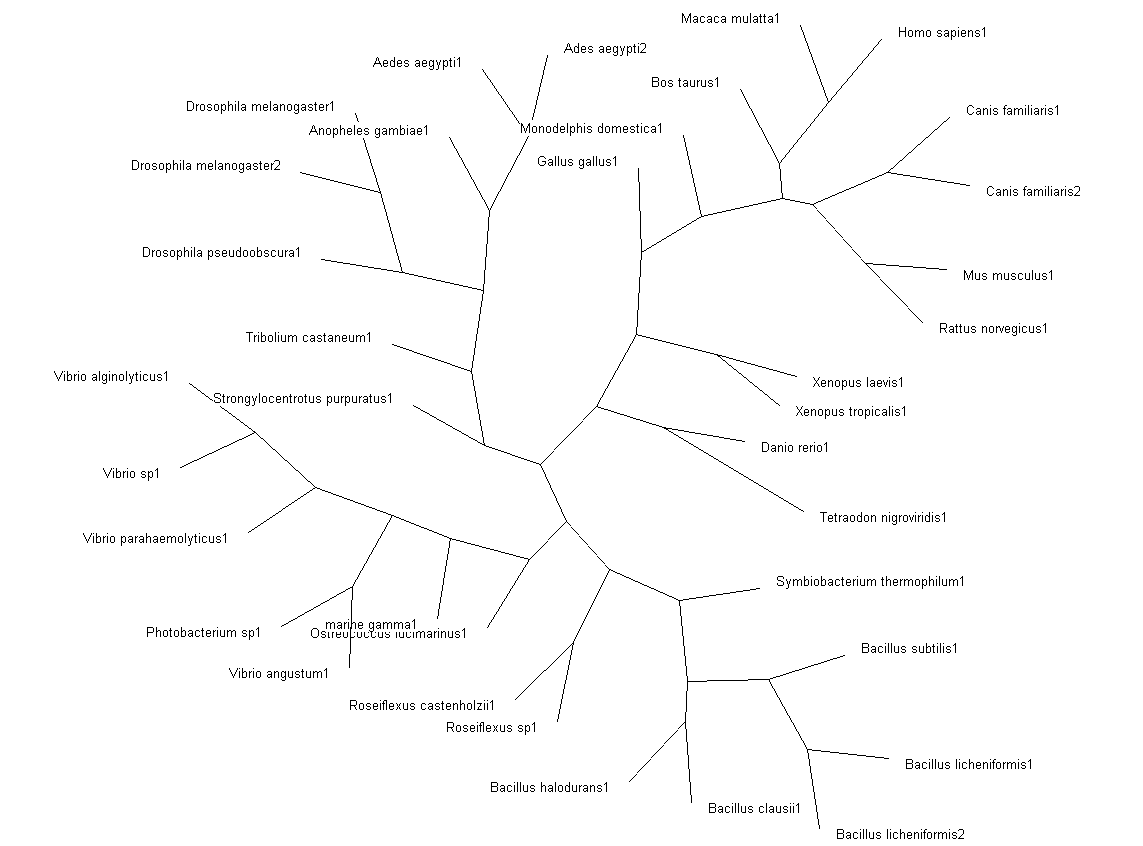
From the Rectangular Cladogram view, it could be observed that there are four distinct separate groups.
From the Rectangular Cladogram view of the tree, it was observed that there were two main Domains — Procaryotes and Eucaryotes. This would also be the root and first branching point of the phylogenetic tree.
In the Eucaryotic part of the tree, the protein could first be observed in Deuterostomia phylum. From there, it was branched off to both Chordata and Echinodermata phyla.
Strongylocentrotus purpuratus (Purple Sea Urchin), forms part of the Echinodermata phylum.
The phylum Chordata would then be further branched into Xenopus laevis (African Clawed Frog) in Amphibia; in Gallus gallus (Red Junglefowl) in Aves; and Mus musculus (House Mouse) in Mammalia classes, respectively.
The Arthropoda phylum, branched off from Deuterostomia, would be further branched into Hexapoda class, consisting winged insects Tribolium castaneum (Rust-red Flour Beetle), Aedes aegypti (Aedes mosquito) and Drosophila melanogaster (Fruit Fly).
For the Procaryotic domain, branching mainly occurs between Gram-positive (Bacillus Sp.) and Gram-negative (Vibrio Sp.) bacteria.
Hence, it can be generally deduced that the N-acetylneuraminic acid (hydrolase) protein is non-evolutionary specific, as it is observed to be present in almost all main phyla and classes of organisms from the two main Procaryotic and Eucaryotic Domains. Its functional significance would therefore be a general one.
Bootstrapping
See Materials and Methods
Bootstrapping values obtained were analysed. Branch values occurring below 75% (<75%) are indicated by an asterisk (*) [Fig. 4].
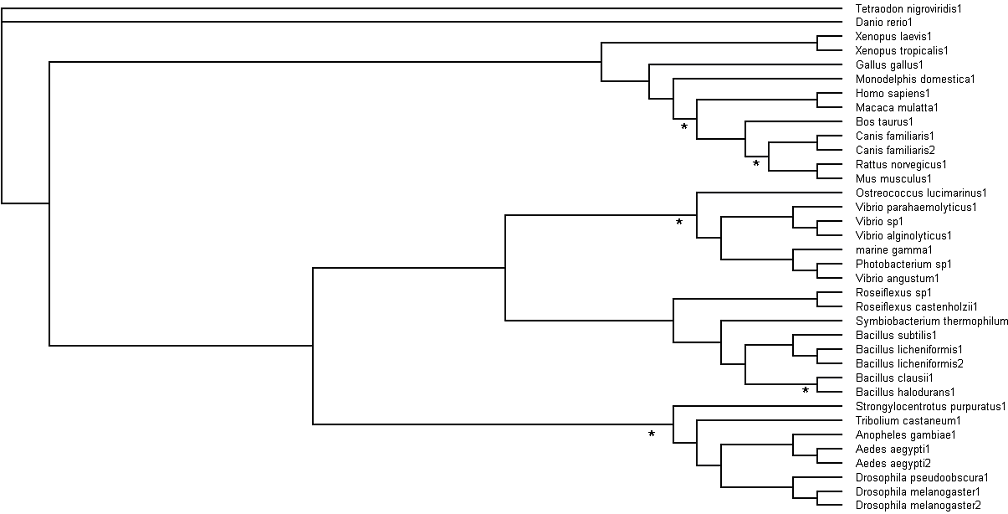
Fig. 4: Branch bootstrap values in Rectangular Cladogram view. Branches with strap values <75% were indicated with asterisks (*)
Tree bootstrapping is necessary to test for the reliability of the branching patterns and distances formed on the phylogenetic tree. This was done by making “pseudoreplicates” of multiple sequence alignments of up to 100 sets. The distance matrices were recalculated using these duplicate alignment values to generate a bootstrap tree, which can be used to compare the branching patterns and distances with the original phylogenetic tree.
The bootstrap values (in percentage) obtained on each branch, signify branching confidence. Bootstrap values of 95% equate to full branching confidence; 75% value equates to 95% branching confidence; 60% value equates to much lowered branching confidence; while 50% value would render no branching confidence.
Proceed to Function
Back to Home Page







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}